Risikomanagement gemäß der europäischen Gesetzgebung und EN ISO 14971:2019
Allgemeiner Hintergrund
Gemäß Anhang I, Abschnitt 3 der Verordnung (EU) 2017/45 (EU-MDR) müssen Hersteller von Medizinprodukten ein Risikomanagementsystem einrichten, umsetzen, dokumentieren und aufrechterhalten, um eine Zulassung für ihre Produkte in Europa zu erhalten. Die harmonisierte Norm EN ISO 14971:2019 definiert die spezifischen Risikomanagementaktivitäten detaillierter als die MDR. Die ISO 14971 bietet den Herstellern einen Rahmen für die Anwendung eines systematischen Ansatzes zur Beherrschung der Risiken, die mit der Verwendung ihrer Medizinprodukte – einschließlich In-vitro-Diagnostika – verbunden sind. Sie wird auch von Regulierungsbehörden außerhalb der EU, einschließlich der USA, Kanada und Australien, offiziell als Risikomanagementnorm anerkannt.
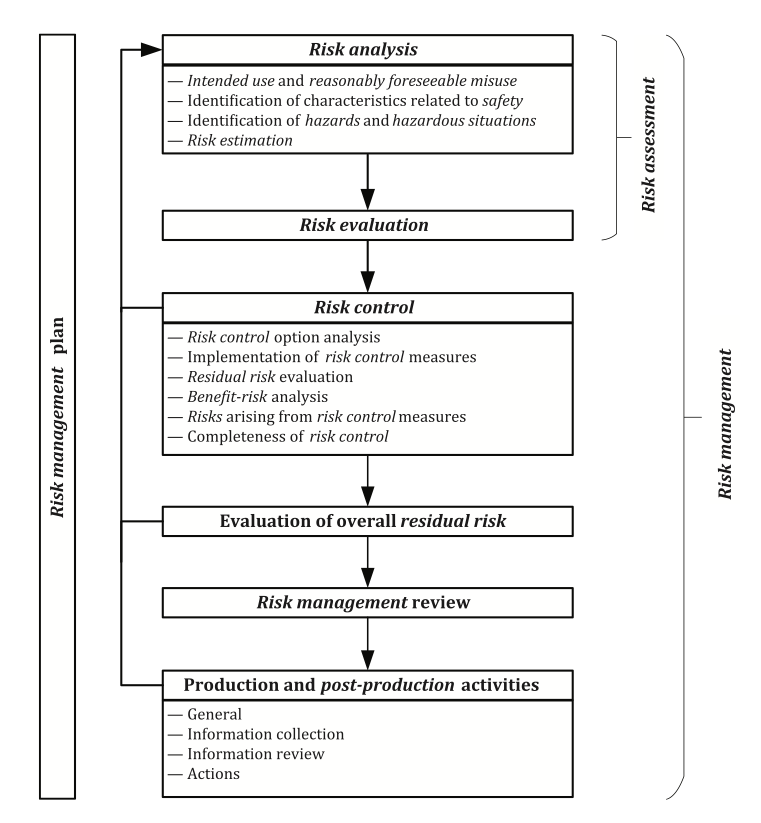
Die ISO 14971 stellt die Anforderungen an einen Risikomanagementprozess als Teil des Risikomanagementsystems des Herstellers in einer schematischen Darstellung dar:
Zu den wichtigsten Überlegungen für Hersteller im Rahmen der EU-IVDR gehören:
Anhand dieser Darstellung wird deutlich, dass alle Risikomanagementaktivitäten geplant werden müssen. Der Risikomanagementplan dokumentiert einen „Fahrplan“ für alle Risikomanagementaktivitäten, die während des gesamten Lebenszyklus des Medizinprodukts durchgeführt werden müssen. Unter Berücksichtigung der Zweckbestimmung des Medizinprodukts legt der Risikomanagementplan die Kriterien für die Risikoakzeptanz fest, die eine objektive Bewertung der Restrisiken im weiteren Verlauf des Prozesses gewährleisten.
Die Risikoabschätzung besteht aus der Risikoanalyse und der Risikobewertung. Eine wesentliche Voraussetzung für die Durchführung der Risikoanalyse ist die klare Beschreibung der Zweckbestimmung, die eine Unterscheidung zwischen dem korrekten Einsatz oder der korrekten Anwendung des Medizinprodukts und seiner Fehlanwendung ermöglicht. Der Hersteller muss die vernünftigerweise vorhersehbare Fehlanwendung definieren und dokumentieren und anschließend im Risikomanagementprozess berücksichtigen. Als vernünftigerweise vorhersehbare Fehlanwendung gilt die absichtliche Verwendung des Medizinprodukts für eine vom Hersteller nicht spezifizierte oder nicht beabsichtigte Zweckbestimmung oder ein unbeabsichtigter Anwendungsfehler.
Bei der Risikoanalyse müssen außerdem die Merkmale des Medizinprodukts ermittelt werden, die seine Sicherheit beeinträchtigen und die mit der Leistung oder dem Funktionsprinzip des Medizinprodukts zusammenhängen können. Der technische Bericht ISO/TR 24971 bietet eine Anleitung zur Anwendung der ISO 14971 und enthält in Anhang A eine umfangreiche Liste von Fragen, die den Hersteller bei der Ermittlung der sicherheitsrelevanten Merkmale unterstützen. Die Sicherheitsmerkmale können qualitativ oder quantitativ sein. Gegebenenfalls ist es notwendig Grenzwerte zu definieren, die eingehalten werden sollten.
Der nächste Schritt ist die Identifizierung der mit dem Medizinprodukt verbundenen Gefahren und die Ermittlung der vernünftigerweise vorhersehbaren Abläufe oder Kombinationen von Ereignissen, die zu Gefahrensituationen führen können. Je nach Abfolge von Ereignissen können – ausgehend von einer Gefahr – unterschiedliche, weitere Gefahrensituationen entstehen. Eine bestimmte Gefahrensituation kann wiederum – je nach individuellen Umständen – zu unterschiedlichen Arten und Schweregraden von Schäden führen, wobei jede Situation ein eigenes Risiko darstellt.
Die Risikoabschätzung für jede der identifizierten Gefahrensituationen erfordert eine Einschätzung der Schwere eines möglichen Schadens und der Wahrscheinlichkeit seines Eintretens. Für die Risikoanalyse gibt es verschiedene Instrumente. Der Hersteller kann dabei eigenverantwortlich entscheiden, welches Instrument für sein Produkt am besten geeignet ist (z. B. vorläufige Gefährdungsanalyse, Fehlerbaumanalyse, Ereignisbaumanalyse, Fehlermöglichkeits- und Einflussanalyse, Gefährdungs- und Beherrschbarkeitsstudie, Gefährdungsanalyse und kritischer Kontrollpunkt).
Die Risikoakzeptanz wird anhand der im Risikomanagementplan definierten Risikoakzeptanzmatrix abgeschätzt. Letztere umfasst die gesamte Risikobewertung. Es müssen alle Gefahrensituationen und alle Arten von möglichen Schäden berücksichtigt werden. Würde man sich nur auf die Worst-Case-Szenarien mit dem höchsten Schadensausmaß konzentrieren, blieben Szenarien mit weniger schwerwiegenden Schäden unberücksichtigt, deren Eintreten wahrscheinlicher ist und die möglicherweise zu einem höheren Risiko führen könnten. Die Schlussfolgerungen der Risikobewertung müssen in der Risikomanagementakte dokumentiert werden. Anhang C.6 der ISO/TR 24971 enthält Beispiele für die Bestimmung der Risikoakzeptanz unter Berücksichtigung verschiedener Elemente und Ansätze für die Risikobewertung: Regulatorische Anforderungen, internationale Normen, Stand der Technik und Bedenken der Interessengruppen. Jedes Risiko, das als akzeptabel eingestuft wird, wird direkt zum Restrisiko. Für jedes Risiko, das als nicht akzeptabel eingestuft wird, müssen Maßnahmen zur Risikokontrolle ergriffen werden.
Es gibt mehrere Optionen der Risikokontrolle, um Risiken zu beseitigen oder auf ein akzeptables Maß zu reduzieren. Sie müssen in der folgenden Reihenfolge durchgeführt werden:
- Eliminierung des Risikos durch eine sichere Auslegung des Medizinprodukts und seines Herstellungsprozesses selbst
- Durchführung von Schutzmaßnahmen bei der Auslegung des Medizinprodukts oder beim Herstellungsprozess, wodurch
- die Wahrscheinlichkeit des Auftretens einer gefährlichen Situation oder eines Schadens verringert wird und/oder
- die Schwere des Schadens verringert wird.
- Bereitstellung von Sicherheitsinformationen für die Anwender des Medizinprodukts (z. B. Warnungen oder Kontraindikationen oder Anweisungen für die Handhabung und Verwendung des Medizinprodukts)
Die ausgewiesenen Risikokontrollmaßnahmen müssen umgesetzt werden. Die Umsetzung sowie die Wirksamkeit der umgesetzten Risikokontrollmaßnahmen müssen überprüft werden. Letztlich sind in der Risikomanagementakte die Ergebnisse dieser Überprüfungen zu dokumentieren.
Ein etwaiges Restrisiko muss nach der Umsetzung der Risikokontrollmaßnahmen anhand der vordefinierten Kriterien für die Risikoakzeptanz erneut evaluiert und bewertet werden. Für jedes Risiko, das als nicht akzeptabel eingestuft wird, müssen in einem iterativen Prozess zwischen Risikokontrolle und Risikoabschätzung weitere Maßnahmen zur Risikokontrolle erwogen werden. Kommt der Hersteller nach eingehender Analyse zu dem Schluss, dass eine weitere Risikominderung nicht möglich ist, kann eine Nutzen-Risiko-Analyse durchgeführt werden.
Nutzen-Risiko-Analyse
Für eine Nutzen-Risiko-Bewertung sind nach ISO/TR 24971 mehrere Aspekte zu berücksichtigen:
- Charakterisierung der Krankheit oder des Zustands der vorgesehenen Patientenzielgruppe
- Unsicherheiten in der Datenlage (eine erste Literaturrecherche zu den Gefahren und dem in Frage kommenden Medizinprodukt kann Aufschluss über das Verhältnis zwischen Nutzen und Risiko geben)
- Informationen aus der Produktion und der, der Herstellung nachgelagerten Phasen ähnlicher, bereits auf dem Markt befindlicher Medizinprodukte
- Der allgemein anerkannte Stand der Technik
- Ein Vergleich des Nutzens des in der Entwicklung befindlichen Medizinprodukts mit dem Nutzen ähnlicher auf dem Markt befindlicher Medizinprodukte
- Ein Vergleich der Restrisiken des in Entwicklung befindlichen Medizinprodukts mit den Restrisiken ähnlicher auf dem Markt befindlicher Medizinprodukte.
Die Sammlung und Analyse von derartigen Daten und Informationen aus der Literatur ermöglicht es festzustellen, ob der Nutzen der Anwendung eines Medizinprodukts dessen Restrisiko überwiegt. Falls dies nicht der Fall ist, muss der Hersteller eine Änderung des Medizinprodukts in Erwägung ziehen, um das spezifische Risiko zu beseitigen. Alternativ kann auch eine Einschränkung des Verwendungszwecks erforderlich sein, z. B. der Ausschluss einer gefährdeten Patientengruppe.
Der Hersteller muss überprüfen, ob alle festgestellten Gefahrensituationen beseitigt und alle Risikokontrollmaßnahmen abgeschlossen wurden. Falls die ausgewählten und umgesetzten Risikokontrollmaßnahmen zu neuen Risiken führen, müssen diese ebenfalls analysiert, bewertet und kontrolliert werden.
Nach ISO 14971 ist es erforderlich, dass der Anteil jedes einzelnen Restrisikos berücksichtigt wird und dass das Gesamtrestrisiko im Verhältnis zum Nutzen des Medizinprodukts bei bestimmungsgemäßem Gebrauch bewertet wird.
Die ISO/TR 24971 enthält eine detailliertere Anleitung zu möglichen Herangehensweisen bei der Risikobewertung. Ebenso zu Inputs und anderen Aspekten, die berücksichtigt werden können. Es ist wichtig zu berücksichtigen, dass sich die Kriterien für die Annehmbarkeit des Gesamtrestrisikos von den Kriterien für die Annehmbarkeit der einzelnen Risiken unterscheiden können. In den Leitlinien wird betont, dass die Kriterien für die Bewertung des Gesamtrestrisikos häufig auf zusätzlichen Elementen beruhen, wie z. B. dem Nutzen der vorgesehenen Verwendung des Medizinprodukts. Der Hersteller ist für die Festlegung einer geeigneten Methode zur Bewertung des Gesamtrestrisikos verantwortlich. Hierzu gibt es keinen „Königsweg“.
Beispiele für die Herangehensweise zur Bewertung des Gesamtrestrisikos:
- Abwägung des Nutzens im Zusammenhang mit der beabsichtigten Anwendung des Medizinprodukts gegenüber dem Gesamtrestrisiko. Dies erfolgt unter Berücksichtigung, dass der Nutzen eindeutig beschrieben und quantifiziert werden kann; die Wahrscheinlichkeit, dass der Nutzen in der vorgesehenen Patientenpopulation wahrgenommen wird; der Dauer und Häufigkeit des Nutzens unter Berücksichtigung der vorgesehenen medizinischen Indikation, des allgemein anerkannten Stands der Technik und Medizin sowie der Verfügbarkeit alternativer Medizinprodukte oder Therapien
- Visuelle Darstellung der Restrisiken
- Vergleich des zu prüfenden Medizinprodukts mit ähnlichen auf dem Markt erhältlichen Medizinprodukten, einschließlich aktueller Informationen über die Zweckbestimmung und die Nebenwirkungen ähnlicher Medizinprodukte sowie Informationen aus der wissenschaftlichen Literatur, einschließlich Informationen über klinische Erfahrungen
- Berücksichtigung von Expertenurteilen zur Unterstützung der Bewertung des Gesamtrestrisikos im Verhältnis zum erwarteten Nutzen bei Anwendung des Medizinprodukts
- Gegebenenfalls weitere Untersuchungen zu bestimmten Risiken (viele Risiken sind möglicherweise nahezu inakzeptabel; dadurch kann das Gesamtrestrisiko inakzeptabel sein, sofern nicht weitere Untersuchungen initiiert wurden)
- Zusätzliche Prüfungen können auch dann erforderlich werden, wenn dezidierte Risiken entweder hinsichtlich ihrer Ursachen oder der angewandten Risikokontrollmaßnahmen voneinander abhängen
Um eine Zulassung für ein Produkt zu erhalten, muss der Hersteller zu dem Schluss kommen, dass das betreffende Medizinprodukt ein positives Nutzen-Risiko-Verhältnis aufweist.
Der Hersteller ist außerdem verpflichtet, die Anwender über alle signifikanten Restrisiken zu informieren und diese Risiken durch entsprechende Angaben in der Begleitdokumentation des Medizinprodukts offenzulegen. Die Offenlegung von Restrisiken unterscheidet sich von den Informationen zur Sicherheit, die eine Maßnahme zur Risikokontrolle darstellen. Grundsätzlich haben Sicherheitsinformationen instruktiven Charakter und sollen dem Anwender Informationen darüber liefern, z. B. wie das Medizinprodukt anzuwenden ist, welche Maßnahmen gegebenenfalls zu ergreifen sind oder wie bestimmte Gefahrensituationen oder eine Entstehung von Schäden vermieden werden können.
Nach der Auslegung und Entwicklung des Medizinprodukts und vor seinem kommerziellen Vertrieb muss der Hersteller überprüfen, ob der Risikomanagementplan ordnungsgemäß ausgeführt und umgesetzt wurde. Mit der Überprüfung des Risikomanagements soll auch sichergestellt werden, dass das Restrisiko insgesamt akzeptabel ist und dass geeignete Methoden zur Erfassung und Überprüfung relevanter Informationen über die Herstellung und über die der Herstellung nachgelagerten Phasen vorhanden sind. Der Risikomanagementbericht wird nach der Überprüfung der Durchführung des Risikomanagementplans erstellt. Der Hersteller muss sicherstellen, dass alle bereitgestellten Informationen in der gesamten Technischen Dokumentation aufeinander abgestimmt und konsistent sind.
Die vom Hersteller erstellte Risikomanagementakte muss mit Informationen aus der Herstellung und der Herstellung nachgelagerten Phasen und aus dem System zur Überwachung nach dem Inverkehrbringen aktualisiert werden. Dabei geht es um eine kontinuierliche Bewertung der Gefahren und der Häufigkeit ihres Auftretens, Abschätzungen der damit verbundenen Risiken sowie des Gesamtrisikos, des Nutzen-Risiko-Verhältnisses und der Risikoakzeptanz. Auf dieser Basis wird eine Schlussfolgerung ermöglicht, ob die Notwendigkeit für Änderungen oder Ergänzungen bezüglich zusätzlicher Kontrollmaßnahmen besteht.
Der Abschnitt über Informationen aus der Herstellung und der, der Herstellung nachgelagerten Phasen aus der ISO 14971 beschreibt die Anforderungen und Aktivitäten in vier Abschnitten. Sie repräsentieren die einzelnen Schritte, die der Hersteller im Rahmen seines Risikomanagements nach Etablierung des RM-Systems durchführen muss (weitere Beispiele sind in der ISO/TR 24971 enthalten):
- Einrichtung eines Systems zur Erfassung und Überprüfung relevanter Informationen aus der Herstellung und der, der Herstellung nachgelagerten Phasen und Festlegung der erforderlichen Aktivitäten im Risikomanagementplan; dazu gehört unter anderem die Festlegung geeigneter Methoden für die Erfassung und Verarbeitung von Daten, z. B. statistische Methoden für die Trendanalyse; es wird vorgeschlagen, die von einem Qualitätsmanagementsystem geforderten Überwachungs- und Feedbackprozesse zu integrieren
- Proaktives Sammeln relevanter Informationen für das zu prüfende Medizinprodukt (z. B. Informationen von Anwendern, aus der Lieferkette und über den allgemein anerkannten Stand der Technik (z. B. neue oder überarbeitete Normen, alternative Medizinprodukte oder alternative Therapien), öffentlich verfügbare Informationen über ähnliche Medizinprodukte und ähnliche andere Produkte auf dem Markt
- Die Überprüfung der gesammelten Informationen ist relevant für die Sicherheit des Medizinprodukts (Feststellung, ob eine bisher nicht erkannte Gefahr oder gefährliche Situation besteht, ein abgeschätztes Risiko nicht mehr akzeptabel ist, der Nutzen des Medizinprodukts das Gesamtrestrisiko nicht mehr überwiegt oder sich der allgemein anerkannte Stand der Technik geändert hat)
- Einleitung notwendiger Maßnahmen im Falle der Identifizierung relevanter Sicherheitsinformationen (einschließlich einer Überprüfung der Risikomanagementakte und der Feststellung, ob ein neues Risiko bewertet werden muss. Ferner, ob ein zuvor bewertetes Risiko erneut bewertet werden muss und ob es gegebenenfalls notwendig ist, zusätzliche Risikokontrollmaßnahmen zu ergreifen; Abschätzung, ob Maßnahmen in Bezug auf bereits auf dem Markt befindliche Medizinprodukte erforderlich sind; Bewertung der Auswirkungen auf zuvor durchgeführte Risikomanagementaktivitäten, die potenziell einen wertvollen Beitrag für die Geschäftsleitung darstellen, wenn diese die Eignung des Risikomanagementprozesses überprüft)
Support & Training
Wenden Sie sich an AKRA TEAM, wenn Sie Unterstützung, praktische Umsetzung und personalisierte Schulungen von Experten mit Schlüsselkompetenzen in den unten aufgeführten Bereichen benötigen.
Kernpunkte
Die Einhaltung der EU-MDR verlangt vom Hersteller die Einrichtung, Umsetzung, Dokumentation und Aufrechterhaltung eines Risikomanagementsystems
Spezifische Risikomanagementaktivitäten sind in der harmonisierten Norm EN ISO 14971:2019 definiert und zusätzliche Anleitungen zur Anwendung der Anforderungen der Norm sind im technischen Bericht ISO/TR 24971 enthalten
Eine klare Beschreibung der Zweckbestimmung ist die Voraussetzung für die Risikoanalyse, da sie eine Unterscheidung zwischen dem korrekten Einsatz oder der korrekten Anwendung des Medizinprodukts und seiner missbräuchlichen Verwendung ermöglicht. Der Verwendungszweck umfasst die medizinische Indikation und Anwendung (Art der Erkrankung, Gewebe und Körperteil), die vorgesehene Behandlungspopulation (Kinder, Erwachsene, ältere Menschen oder spezielle Patientengruppen wie Schwangere und Stillende), die Anwender und das Anwendungsumfeld (Laienanwender zu Hause, professionelle Anwender in oder außerhalb einer klinischen Umgebung) sowie das Funktionsprinzip (d.h. wie die Diagnose oder Behandlung erreicht wird).
Der Risikomanagementplan muss die Methoden spezifizieren, die zur Untersuchung der qualitativen und quantitativen Aspekte der klinischen Sicherheit angewandt werden. Dazu gehört eine eingehende Erläuterung, wie Restrisiken und Nebenwirkungen bestimmt werden.
Das Gesamtrestrisiko eines Medizinprodukts muss akzeptabel sein, und der Hersteller muss in der Lage sein, angemessen zu begründen, dass das Nutzen-Risiko-Verhältnis des Medizinprodukts positiv ist.
Unsere Leistungen
Auch wenn die harmonisierte Norm einen Rahmen für den Risikomanagementprozess vorgibt, verbleiben viele Herausforderungen bei der Umsetzung der einzelnen Prozessschritte für ein bestimmtes Medizinprodukt. AKRA TEAM kann Sie mit pragmatischen Ansätzen unterstützen, die gesetzlichen Vorgaben einzuhalten. Dadurch erhöhen Sie die Wahrscheinlichkeit für eine erfolgreiche Zertifizierung.
Schulungen
AKRA TEAM bietet Schulungen zur Einführung und Aufrechterhaltung eines Risikomanagementsystems gemäß den Anforderungen der EU-MDR und der harmonisierten Norm EN ISO 14971:2019 an. Im Rahmen dieses Trainings wird das erforderliche Grundlagenwissen vermittelt, welches Ihre Mitarbeiter im Rahmen der Risikomanagementaufgaben benötigen.
Entwicklung von Verfahren und Vorlagen Entwicklung von Verfahrensanweisungen und Dokumentvorlagen
AKRA TEAM kann Sie bei der Erstellung der erforderlichen Verfahrensanweisungen für Ihren Risikomanagementprozess unterstützen, einschließlich der Definition von Aktivitäten nach der Herstellung. Ferner mit Dokumentvorlagen für Risikomanagementpläne und Risikomanagementberichte.
Gap-Analysen
AKRA TEAM bietet umfassende Gap-Analysen der Risikomanagementakte an. In diesen Analysen werden Risiken identifiziert und Verbesserungsvorschläge erarbeitet. Im Rahmen der Gap-Analyse werden zunächst potenzielle Risiken für die Einreichung identifiziert (von gering bis hoch). Im folgenden Schritt schlägt AKRA TEAM Ihnen Maßnahmen zur Risikominderung und Optimierung der Dokumente vor. So können Sie Korrekturen vornehmen, bevor Sie die Dokumente zur Prüfung durch die benannte Stelle einreichen. Dies umfasst eine Überprüfung von allen Teilen der Risikomanagementakte, insbesondere des Risikomanagementprozesses, des Risikomanagementplans und -Berichts sowie der Risikoanalyse einschließlich der Risikoeinschätzung und der Risikokontrollmaßnahmen, des Ergebnisses der Nutzen-Risiko-Bewertung und der Vollständigkeit der offenzulegenden Restrisiken.
Umsetzung
AKRA TEAM kann Sie bei der Umsetzung Ihrer Risikomanagementaktivitäten unterstützen um sicherzustellen, dass der Risikomanagementplan ordnungsgemäß umgesetzt wird und dass alle Informationen innerhalb der Technischen Dokumentation konsistent und miteinander abgeglichen sind. Dadurch wird zudem sichergestellt, dass alle in der klinischen Bewertung ermittelten klinischen Risiken in der Risikodatei erfasst werden.
Kontinuierliche Aktualisierung der Dokumentation
AKRA TEAM kann Sie bei der Aktualisierung der Dokumentation entsprechend den Informationen aus der Produktion und aus dem System zur Überwachung nach dem Inverkehrbringen (PMS) unterstützen. Dazu gehört z.B: die Verbesserung von Prozessen, mit Hilfe derer die kontinuierliche Aktualisierung und Bewertung der Gefahren und ihrer Häufigkeit erleichtert wird. Ebenso umfasst dies eine Erleichterung der Bewertung der damit verbundenen Risiken sowie des Gesamtrisikos, des Nutzen-Risiko-Verhältnisses und der Risikoakzeptanz insgesamt.