Kombinationsprodukte
Allgemeiner Hintergrund
Kombinationsprodukte (Drug-Device-Combinations, DDCs) stellen eine heterogene Gruppe von Produkten dar, die eine Kombination aus Medizinprodukten einerseits und Arzneimitteln andererseits sind. Diese Produktgruppe hat sich in den letzten Jahren erheblich weiterentwickelt, da sie ein hohes Potenzial an klinischem Nutzen für die Behandlung verschiedener Arten von Krankheiten birgt. Unter anderem können DDCs Patienten zugutekommen, die ihre Medikamente regelmäßig und langfristig ambulant einnehmen müssen, entweder durch Selbstmedikation oder mit Unterstützung eines professionellen Anwenders. Dies verringert die Belastung der Patienten und der Gesundheitssysteme und fördert die Sicherheit der Medikamenteneinnahme durch die Patienten.
Das Produktspektrum reicht von einfachen vorgefüllten Spritzen (die mit einer Arzneimittelkomponente gefüllt sind) über automatische Injektionsgeräte bis hin zu komplexen „intelligenten“ Systemen zur bedarfsgerechten Arzneimittelabgabe, die implantiert werden (z. B. Insulinpumpen). Bei anderen Geräten handelt es sich um eine Kombination aus einem herkömmlichen Medizinprodukt (z. B. einem Stent), das mit einem Arzneimittel beschichtet ist, um das klinische Ergebnis der Behandlung zu verbessern (z. B. beschichteter Stent).
Nomenklatur und geltende Rechtsvorschriften
Bei DDCs wird immer ein Medizinprodukt mit einem Arzneimittel kombiniert, was dazu führt, dass zwei verschiedene Rechtsvorschriften anwendbar sind. In Europa sind dies zum einen die EU-Verordnung über Medizinprodukte (Verordnung 2017/745) und zum anderen die Richtlinie über Medizinprodukte (Richtlinie 2001/83/EG).
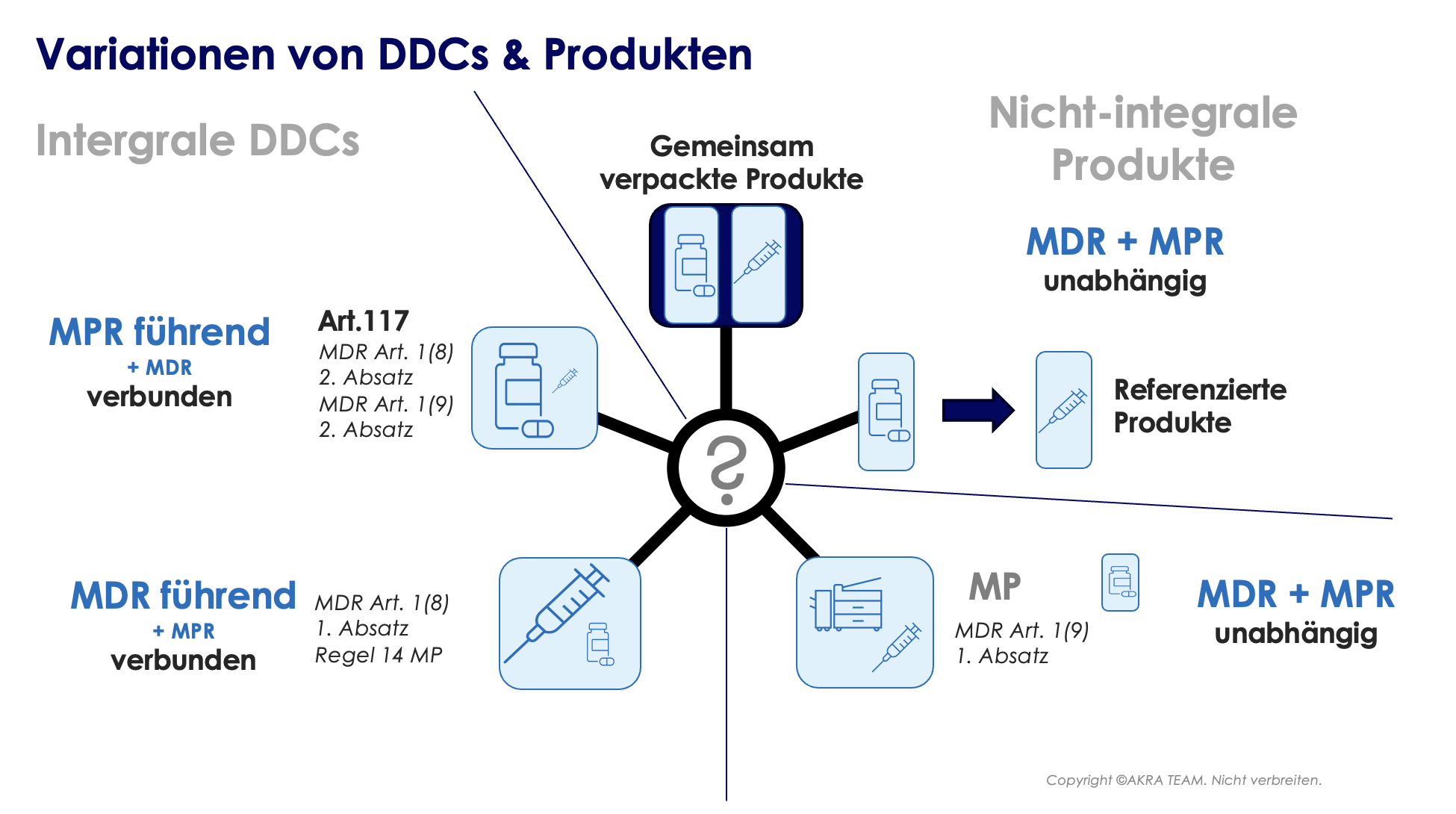
Die Nomenklatur der Kombinationsprodukte ist in Europa noch nicht einheitlich; im Wesentlichen wird oft zwischen integralen und nicht-integralen Kombinationsprodukten unterschieden.
Nicht integrale DDCs: Es gibt Fälle, in denen ein Arzneimittel und ein Medizinprodukt in derselben Sekundärverpackung (= Mitverpackung/Beilegen von Produkten) in Verkehr gebracht werden, aber kein integrales Produkt bilden. Z.B. die Kombination von einem Flacon, gefüllt mit einem Arzneimittel, mit einer (leeren) sterilen Spritze mit CE-Kennzeichnung. Dieses Produkt wird NICHT als Kombinationsprodukt betrachtet, da das Medizinprodukt unter Artikel 1(9) Absatz 1 der EU-MDR fällt.
Darüber hinaus gelten Produkte, auf die in der Arzneimittelinformation verwiesen wird, oder Arzneimittel, auf die in den mit dem Produkt gelieferten Informationen verwiesen wird, NICHT als DDCs. Die Anforderungen für diese Produkte werden in Abschnitt 3 der Fragen und Antworten des Leitfadens EMA/37991/2019 behandelt. Bei Zweifeln, ob es sich bei einem bestimmten Produkt um ein Kombinationsprodukt handelt, empfiehlt es sich, die MDCG 2022-5 zu konsultieren („Guidance on Borderline“).
Unabhängig von der Nomenklatur ist die anwendbare „führende“ Gesetzgebung immer eindeutig und hängt von der Hauptwirkung des Produkts ab. Sofern die Hauptwirkung vom Medizinproduktteil bewerkstelligt wird, ist die EU-MDR die primär anzuwendende Gesetzgebung. Im anderen Fall ist die Medizinprodukte-Richtlinie (MPR = Richtlinie 2001/83/EG) die primär anwendbare Gesetzgebung (siehe Abbildung unten).
Integrale DDCs: Im Folgenden werden 2 Produktgruppen genauer betrachtet:
- Integrale DDCs, bei denen vom Medizinprodukt (MP) die primäre Wirkung ausgeht
- Integrale DDCs, bei denen von der pharmazeutischen Komponente (PK) die primäre Wirkung ausgeht
Integrale DDCs / primäre Wirkung durch das MP
Beispiele: Produkte, die ein integriertes ergänzendes Arzneimittel enthalten
- Mit Heparin oder einem Antibiotikum beschichtete Katheter
- Antibiotikahaltige Knochenzemente
- Wurzelkanalfüller, die Arzneimittel enthalten, deren Wirkung mit der des MP zusammenhängt
- Weichgewebefüller mit Lokalanästhetika
- Wachstumsfaktoren enthaltende Knochenfüllmaterialien
- Mit Spermiziden beschichtete Kondome
- Elektroden mit steroidbeschichteter Spitze
- Wundverbände, chirurgische Abdeckungen oder Barriereabdeckungen (einschließlich Tüllverbänden) mit antimikrobiellem Wirkstoff
- Intrauterine Verhütungsmittel, die Kupfer oder Silber enthalten
- Ophthalmische Spüllösungen, die hauptsächlich zur Spülung bestimmt sind und Bestandteile enthalten, die den Stoffwechsel der Endothelzellen der Hornhaut unterstützen
- Mit Arzneimitteln beschichtete Koronarstents
- Blutbeutel, die Stoffe enthalten, die bei getrennter Verwendung als Arzneimittel gelten würden
- Flüssiger Wundverband mit antimikrobiellem Wirkstoff
Quelle: MDCG 2022-5
Integrale DDCs / primäre Wirkung durch das Arzneimittel
Beispiele für integrierte DDCs
- Mit einem Arzneimittel vorgefüllte Spritzen
- Aerosole, die ein Arzneimittel enthalten
- Vernebler, die mit einem bestimmten Arzneimittel vorgeladen sind
- Pflaster für die transdermale Verabreichung von Medikamenten
- Implantate, die Arzneimittel in einer Polymermatrix enthalten, deren Zweck die Freisetzung des Arzneimittels ist, z. B. Kunststoffkügelchen, die ein Antibiotikum zur Behandlung von Knocheninfektionen enthalten, oder eine Matrix zur Freisetzung osteoinduktiver Proteine in den umgebenden Knochen
- Intrauterine Empfängnisverhütungsmittel, die Gestagene abgeben sollen
- Einweg-Iontophoresegeräte, die ein Arzneimittel enthalten und dazu bestimmt sind, das Arzneimittel zur Behandlung einer Erkrankung abzugeben
- Wundbehandlungsprodukte, die eine Matrix umfassen, deren Zweck die Verabreichung von Arzneimitteln ist, z. B. Wundverbände, die ein antimikrobielles Mittel enthalten, wobei die primäre Wirkung des Verbandes darin besteht, das Mittel der Wunde zum Zweck der Infektionskontrolle zuzuführen
- Temporäre Wurzelkanalfüller, die Arzneimittel enthalten und deren Zweck die Abgabe des Arzneimittels ist
- Tabletten, die ein Arzneimittel mit eingebettetem Sensor zur Überwachung der Therapietreue enthalten
Quelle: MDCG 2022-5
Eine Neuerung in der EU-MDR repräsentiert Artikel 117. Artikel 117 stellt eine Änderung der Richtlinie 2001/83/EG dar. Arzneimittelhersteller müssen nunmehr bei der Einreichung des Zulassungsdossiers gegenüber der zuständigen Behörde nachweisen, dass der MP-Teil des Kombinationsproduktes mit den geltenden Grundlegenden Leistungs- und Sicherheitsanforderungen (GSPR) gemäß Anhang I der EU-MDR übereinstimmt.
Es bestehen folgende Optionen:
- Der Hersteller stellt eine EU-Konformitätserklärung aus, oder
- Die von einer benannten Stelle ausgestellte Bescheinigung, die den Hersteller berechtigt, das Medizinprodukt mit der CE-Kennzeichnung zu versehen, oder
- Legt die Stellungnahme einer benannten Stelle zum MP-Teil vor.
Sofern der MP-Teil keine CE-Kennzeichnung trägt, ist der letztgenannte Weg für Pharmaunternehmen am wenigsten aufwändig. Die Zusammenarbeit mit benannten Stellen kann jedoch für einen Arzneimittelhersteller neu sein und unterscheidet sich von der Zusammenarbeit mit den zuständigen medizinischen Behörden.
Die daraus resultierenden Herausforderungen reichen von „Wie finde ich eine benannte Stelle“, über die Zusammenarbeit mit einer benannten Stelle (inkl. Fristen) bis hin zu Fragen zur Art, zum Umfang und dem Format der einzureichenden Dokumente.
AKRA TEAM kann Sie in all diesen Phasen unterstützen und Ihnen den Weg durch die Prozesse erleichtern.
Support & Training
Wenden Sie sich an AKRA TEAM, wenn Sie Unterstützung, praktische Umsetzung und personalisierte Schulungen von Experten mit Schlüsselkompetenzen in den unten aufgeführten Bereichen benötigen.
Kernpunkte
Kombinationsprodukte stellen ein stark wachsendes Segment in der Gesundheitsbranche dar. Dies Segment umfasst eine heterogene Gruppe unterschiedlicher Produkte, die ein Medizinprodukt mit einem Arzneimittel kombinieren.
Somit sind beide Rechtsvorschriften, die EU-MDR und die Arzneimittelrichtlinie, anwendbar, wobei sich die maßgebliche Rechtsvorschrift danach richtet, von welchem Part (Medizinprodukt oder Arzneimittel) die Hauptwirkung des DDCs ausgeht.
Insbesondere Artikel 117 der EU-MDR führt zu erheblichen Veränderungen in der regulatorischen Landschaft verschiedener Interessengruppen und erfordert die Anpassung bereits etablierter Prozesse. Bei der Einreichung eines Zulassungsdossiers für Kombinationsprodukte, deren primäre Wirkung auf dem Arzneimittel basiert, muss die Einhaltung der geltenden Grundlegenden Leistungs- und Sicherheitsanforderungen gemäß Anhang I EU-MDR nachgewiesen werden. Im Gegensatz zu früher ist eine benannte Stelle einzubeziehen, sofern der spezifische Medizinprodukte-Part nicht mit einer CE-Kennzeichnung versehen ist, die durch eine Konformitätserklärung und/oder ein Zertifikat einer benannten Stelle belegt wird.
AKRA TEAM kann Sie unterstützen, diesen Prozess effektiv und effizient zu durchlaufen.
Unsere Leistungen
Auswahl einer benannten Stelle für den Medizinprodukt-Teil des Kombinationsproduktes.
Unterstützung der Kommunikation mit der benannten Stelle.
Beratung vor Einreichung der Unterlagen bei der benannten Stelle.
Erstellung von Unterlagen zur Erfüllung der Verpflichtungen nach Artikel 117 EU-MDR.
Gap-Assessment / Readiness-Check von Dokumenten einschließlich detaillierter Empfehlungen zur Einhaltung der Vorschriften.
Beratung bei von der benannten Stelle beanstandeten Nichtkonformitäten/Mängeln. Unterstützung bei Antwortschreiben an die benannte Stelle.
Schulungen zu Kombinationsprodukten (Überblick / Prozesse).