Klinische Prüfungen gemäß EU-MDR
Was wird benötigt für eine gute Strategie zur klinischen Prüfung?
Allgemeiner Hintergrund
Klinische Prüfungen im Rahmen der EU-Medizinprodukteverordnung (EU-MDR) umfassen den Prozess der Durchführung klinischer Prüfungen zur Sammlung von Daten über die Sicherheit und Leistungsfähigkeit von Medizinprodukten, bevor diese auf den Markt gebracht werden. Dies umfasst vor allem auch Fälle, in denen Lücken bezüglich klinischer Nachweise bestehen, wenn z.B. wesentliche Änderungen am Medizinprodukt vorgenommen wurden sowie auch für die erstmalige CE-Kennzeichnung nach EU-MDR. Klinische Studien sind ein wichtiger Teil der regulatorischen Verfahren in der EU und werden für bestimmte Klassen von Medizinprodukten verlangt, um sicherzustellen, dass sie die einschlägigen Sicherheits- und Leistungsstandards erfüllen.
Mit der EU-MDR sind klinische Prüfungen wichtiger denn je. Insbesondere da eine Konformitätsbewertung auf der Grundlage eines Äquivalenzvergleichs oft nicht mehr möglich ist, müssen die Hersteller von Medizinprodukten für eine erfolgreiche Konformitätsbewertung klinische Daten zu ihren eigenen Produkten generieren.
Bei Produkten der Klasse III und implantierbaren Produkten sollten die Daten im Allgemeinen aus klinischen Prüfungen stammen, die in Verantwortung des Sponsors (Herstellers) durchgeführt wurden. Dies gilt auch für Produkte niedrigerer Risikoklassen, die eine neue Zweckbestimmung haben oder auf neuen Technologien basieren, welche Sicherheits- oder andere Bedenken aufwerfen. Die EU-MDR verlangt die Durchführung klinischer Prüfungen für alle die Produkte – unabhängig von ihrer Klassifizierung – bei denen Sicherheitsbedenken bestehen oder die einen hohen Innovationsgrad aufweisen.
Vor Durchführung einer klinischen Prüfung muss der Sponsor der Studie die Genehmigung der zuständigen Behörde des EU-Mitgliedstaats einholen, in dem die Prüfung durchgeführt werden soll. Der Sponsor muss außerdem die Genehmigung einer Ethikkommission einholen. Die Aufgabe der Ethikkommission besteht in der Überprüfung des Studienprotokolls im Hinblick darauf, dass es ethisch einwandfrei ist und die Rechte und die Sicherheit der Studienteilnehmer geschützt werden.
Klinische Prüfungen im Rahmen der EU-MDR müssen in Übereinstimmung mit den Leitlinien der „Guten Klinischen Praxis“ (Good Clinical Practice, GCP) durchgeführt werden, die eine Reihe von internationalen Standards für die Planung, Durchführung und Berichterstattung von klinischen Prüfungen enthalten. Die Leitlinien sollen sicherstellen, dass klinische Prüfungen so durchgeführt werden, dass die Rechte, die Sicherheit und das Wohlergehen der Studienteilnehmer geschützt werden. Ferner, dass verlässliche Daten gewonnen werden können, um regulatorische Fragen zielgerichtet zu beantworten.
Nach Abschluss einer klinischen Prüfung muss der Sponsor einen Bericht über die klinische Prüfung (Clinical Investigation Report, CIR) erstellen, der die Ergebnisse der Studie zusammenfasst und Schlussfolgerungen zur Sicherheit und Leistungsfähigkeit des Medizinprodukts enthält. Der CIR wird dann der zuständigen Behörde zur Prüfung im Rahmen des Konformitätsbewertungsverfahrens vorgelegt.
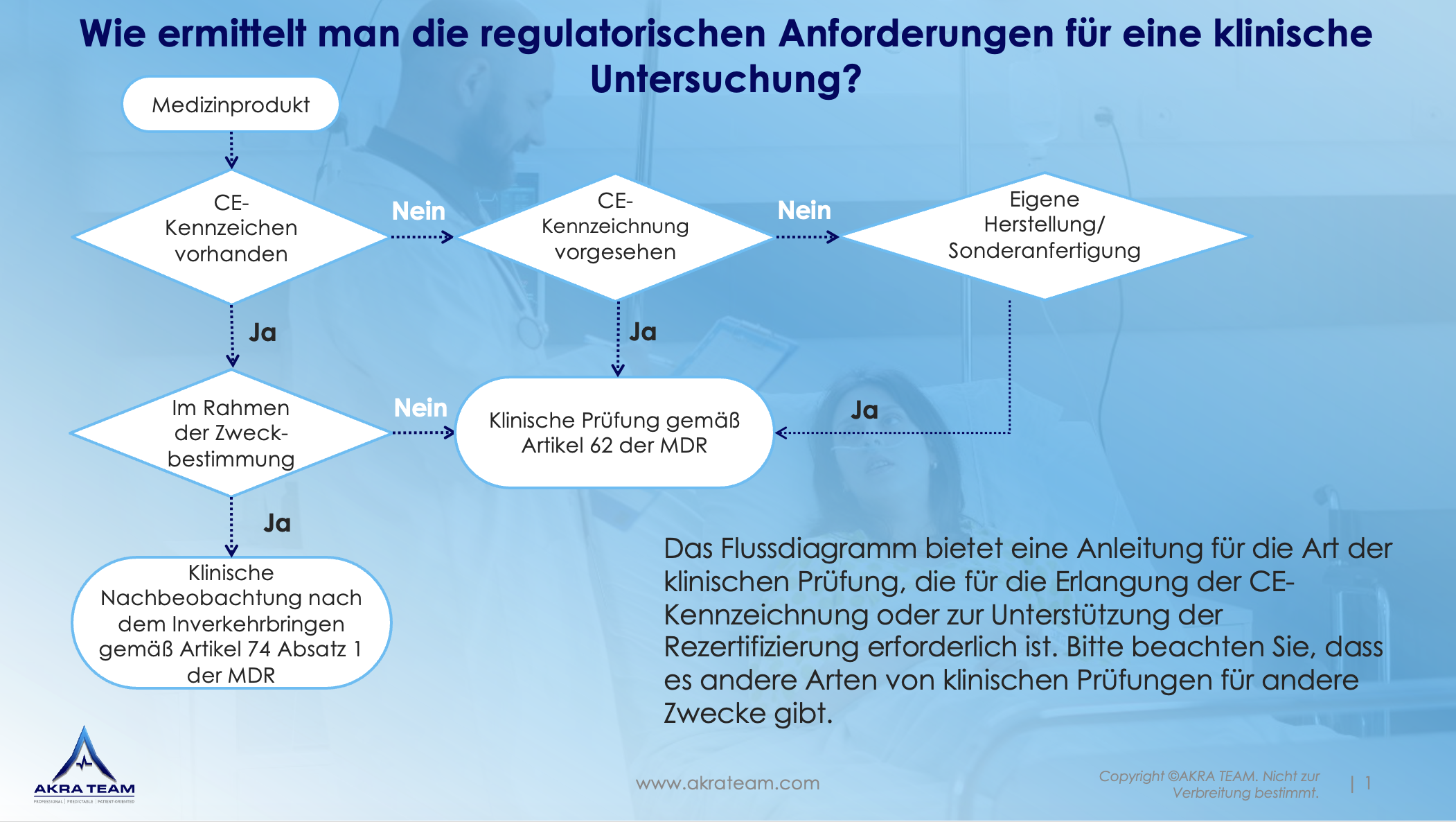
Die Planung und Durchführung einer klinischen Prüfung ist dementsprechend oft ressourcenintensiv. Je nach Art der klinischen Prüfung müssen unterschiedliche Anforderungen erfüllt werden (siehe Flussdiagramm). AKRA TEAM kann Sie dabei unterstützen, klinische Prüfungen effizient und zielgerichtet zu gestalten und zu planen.
Support & Training
Wenden Sie sich an AKRA TEAM, wenn Sie Unterstützung, praktische Umsetzung und personalisierte Schulungen von Experten mit Schlüsselkompetenzen in den unten aufgeführten Bereichen benötigen.
Wichtige Überlegungen:
Stimmen Sie Ihre Strategie der klinischen Prüfung mit Ihrer Marketingstrategie ab: Welche Produktaussagen wollen Sie machen und welche Daten unterstützen diese?
Generieren Sie ausreichend Daten, um das Vertrauen Ihrer Zielgruppen in die Sicherheit und Leistungsfähigkeit Ihres Produkts zu gewinnen.
Berücksichtigen Sie bei der Planung der Ergebnisparameter Ihrer klinischen Prüfung unbedingt Ihre Risikomanagementdaten.
Berücksichtigen Sie die Erstattung durch die Krankenkassen für Ihr Produkt in Ihrem Markt sowie auch die gesetzlichen Anforderungen in anderen potenziellen Zielmärkten.
Vergewissern Sie sich, dass Ihr QMS-System Ihre klinische Prüfung abdeckt, auch wenn einige Tätigkeiten ausgelagert sind.
Wenden Sie die länderspezifischen Anforderungen für die Beantragung und Dokumentation klinischer Prüfungen an.
Unsere Leistungen
Schulungen
AKRA TEAM bietet Schulungen zu den allgemeinen regulatorischen Anforderungen für klinische Prüfungen nach EU-MDR und den entsprechenden Leitfäden an, die von der Medical Device Coordination Group (MDCG) herausgegeben werden.
AKRA TEAM kann außerdem Schulungen zur ISO 14155 „Klinische Prüfung von Medizinprodukten an Menschen - Gute klinische Praxis“ anbieten.
Entwicklung von Verfahrensanweisungen und Dokumentvorlagen
AKRA TEAM kann bei der Erstellung von Verfahrensanweisungen für die Planung und Durchführung klinischer Prüfungen unterstützen, mit entsprechenden Querverweisen zu den korrespondierenden Verfahrensanweisungen des QMS (z. B. Plan zur Durchführung der klinischen Bewertung, CEP). Ebenso kann AKRA TEAM weitere Dokumentvorlagen für klinische Prüfungen bereitstellen.
Gap-Analysen
AKRA TEAM kann eine Gap-Analyse Ihrer vorhandenen Daten und/oder Ihres Plans für klinische Untersuchungen durchführen, um fehlende Informationen zu ermitteln und Hilfestellung für den besten Ansatz zur Schließung der Lücken zu geben.
AKRA TEAM unterstützt Ihr Unternehmen bei der Identifizierung der am besten geeigneten Strategie für die Planung und Durchführung klinischer Studien zu Ihrem Produkt.
AKRA TEAM stellt sicher, dass das Design Ihrer klinischen Prüfung den etablierten internationalen Leitfäden entspricht, wie z. B. der internationalen Norm ISO 14155 „Klinische Prüfung von Medizinprodukten an Menschen - Gute klinische Praxis“ sowie den ethischen Grundsätzen, die in der neuesten Version der „Deklaration von Helsinki“ des Weltärztebundes gemäß MDR Artikel 1 (64) festgelegt sind.
Umsetzung
AKRA TEAM kann Ihnen Feedback z.B. zur Strategie der klinischen Studie, zur Protokollentwicklung, zum CRF-Design und zur Gestaltung der Einverständniserklärung geben. Wir unterstützen Sie darüber hinaus bei der Einreichung von Studien bei den zuständigen Behörden und Ethik-Kommissionen, bei der Vorbereitung des Studienstarts am Studienzentrum sowie dem Beginn und dem Abschluss der Studie.