Clinical Evaluation under EU MDR
General Background
Clinical evaluation is a systematic and planned process to continuously generate, collect, analyze & assess the clinical data pertaining to a device in order to demonstrate the safety and performance, including clinical benefits, of the device when used as intended. Clinical evaluation is required under Article 61 and Annex XIV of the European Medical Device Regulation 2017/745 (EU MDR).
This process involves evaluating the available clinical evidence, such as clinical studies, clinical literature and pre-clinical data to demonstrate that the device is safe and effective for its intended use in line with the generally recognized state of the art (SOTA). The clinical evaluation report is an output that summarizes the results of the clinical evaluation process and is used to support the device’s conformity with the EU MDR. The report must be updated regularly throughout the device’s lifecycle to ensure continued compliance with the EU MDR and forms part of the holistic technical documentation for the device.
The clinical evaluation process typically includes the following key documents:
This documentation includes:
- Clinical evaluation procedure
- Clinical evaluation plan (CEP)
- Literature search protocol & report for SOTA and device and
- Clinical evaluation report (CER)
The clinical evaluation procedure describes the relevant processes and responsibilities defined by the manufacturer to meet the expectations of EU MDR and relevant state of the art guidance.
To conduct clinical evaluation, the following steps can be taken:
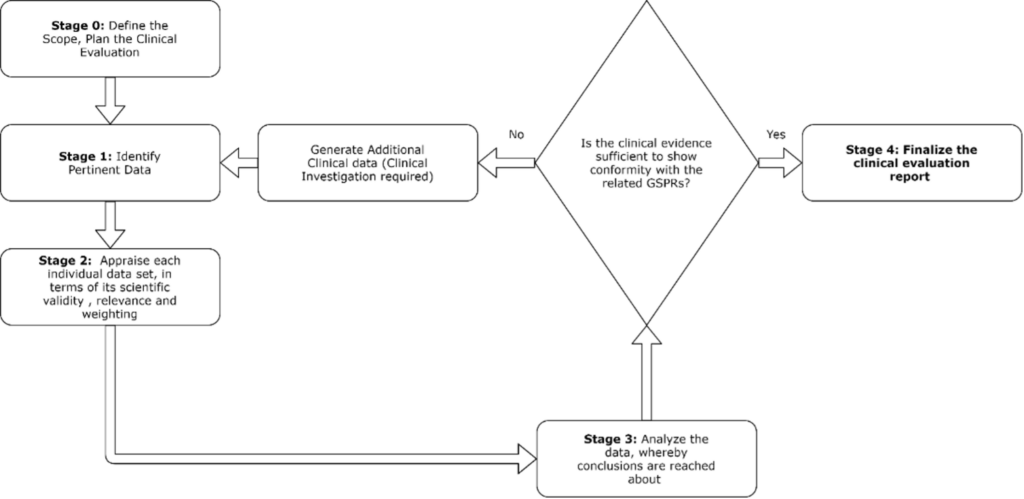
The clinical evaluation plan serves as the basis for the clinical evaluation report, describing the relevant data inputs and document outputs expected.
The clinical evaluation plan includes the following:
- Relevant general safety and performance requirements (GSPRs) that require support from relevant clinical data
- Intended purpose statement
- Intended target patient groups with clear
- Indications and contra-indications
- Intended clinical benefits to patients including measurable safety and performance parameters
- Methods to be used to evaluate qualitative and quantitative aspects of clinical safety with clear reference to the determination of residual risks and side-effects
- List of parameters to be used to determine, based on the state of the art in medicine, the acceptability of the benefit-risk ratio for each indication and for the intended purpose or purpose(s) of the device
- Description of how benefit-risk issues relating to specific components such as use of pharmaceutical, non-viable animal or human tissues, are to be addressed
- A clinical development plan
Stage 0-3 are summarized in the CER and form the basis of the evidence used to demonstrate compliance with the relevant GSPRs.
Clinical evidence is identified from a number of sources. Data relevant to the clinical evaluation may be held by the manufacturer or a third party, or be available in the scientific literature, for the device in question or for comparable devices. All relevant clinical data must be identified for the subject device.
Clinical data is identified based on the strategy proposed in the clinical evaluation plan. Specifically clinical literature is identified by the literature search protocol which includes the relevant literature databases, search terms and inclusion/exclusion criteria.
In the analysis section, manufacturers should provide a brief summary of the data appraisal methods, this includes a description of how data from a given study or other source of data is determined as sufficient quality and relevance to be included in the clinical evaluation. Appraisal also includes evaluation of criteria including study design, sources of bias, peer review, relevance to subject device.
Relevant data sets should be weighted on the basis of scientific quality and relevance to the scope and objectives of the clinical evaluation for the subject devices. The acceptability of the appraisal should be in terms of:
- Methodological quality and scientific validity of articles retrieved and evaluated appropriately
- Relevance of the information to the clinical evaluation determined and documented
- Contribution of each data set to the clinical evaluation weighted according to systematic criteria
The goal of the analysis stage is to provide conclusions for the sufficiency of clinical data in the CER to address the relevant GSPRs to the subject device. Additionally clinical claims are supported with evidence and the benefit/risk determination is presented.
Conclusions will be made regarding the sufficiency of appraised data sets available for the subject device to collectively demonstrate the safety, clinical performance and/or effectiveness of the device in relation to its intended use.
Benefit-risk conclusions should include:
- Summary of the clinical benefits in relation to the meaningful and measurable patient relevant clinical outcome. Their positive impact on patient management or public health.
- Risks with clinical relevance (e.g., uncertainties or limitations of clinical data, undesirable side-effects, potential for misuse, etc.) and provide a short description (e.g. incidence, severity, duration, vulnerable patient subgroups, dose-response relationship where relevant, etc.).
- Impact of risks in relation to the clinical benefits taking into account the factors described and in particular the uncertainties in relation to available clinical data.
Depending on the device under evaluation and clinical strategy, the CER will be structured accordingly. In general, the CER includes the following sections:
- Executive summary
- Scope
- State of the art
- Evidence supporting the device under evaluation.
- Pre-clinical data.
- Clinical literature review.
- Clinical investigation data.
- Post-market surveillance and post-market clinical follow-up data.
- Data appraisal
- Data analysis
- Conclusions
The CER must be evaluated by a person or team with qualifications (relevant university degree and 5 years’ experience or 10 years’ experience) in the field of:
- Research methodology
- Information management
- Regulatory requirements
- Medical writing
- Device technology and its application
- Diagnosis and management of the conditions intended to be diagnosed or managed by
- The device, knowledge of medical alternatives, treatment standards and technology (clinical background)
The CER must undergo a defined periodic review and update to ensure that it remains current and reflects any new information or data that becomes available. This review frequency is dependent upon:
- Whether the device carries significant risks
- Whether the device is well established, taking into consideration:
- Innovation
- Relevant changes in clinical sciences, materials sciences or other sciences related to the device under evaluation
- The current level of confidence in the evaluation of clinical performance and clinical safety of the device
- The total number of devices used so far in the market, and expected reporting rates under the vigilance system
- Whether there are risks and uncertainties or unanswered questions, in the medium or L-term, that would influence the frequency of updates
- Design changes or changes to manufacturing procedures
Support & Training
Contact AKRA TEAM for support, hands on implementation services and personalized training by experts with key competencies in the areas listed below.
Key points
State of the art (SOTA) is heavily scrutinized during notified body review. A thorough review of the clinical background, medical conditions, guidelines/standards, current medical practice, alternative therapies, similar devices, benchmark devices, SOTA benefit-risk analysis and benchmarks of safety and performance are expected to be provided.
Clinical claims for the subject device must be specified in the CER and supported with sufficient clinical data.
Clinical benefits shall be defined for every device and linked to appropriate measurable performance outcomes. The clinical benefit should reflect the intended purpose of the device.
Safety and performance outcomes should be quantitative and patient relevant established from benchmark values obtained from the state of the art literature review.
Choosing the correct clinical strategy is key under EU MDR. Whether the device can claim equivalence, well-established technology, clinical data not deemed appropriate (Art 61(10), or sufficient. The subject device will dictate the quality and quantity of clinical evidence required.
Our Services
Training
AKRA TEAM provides training regarding general Clinical evaluation and CER writing as well as specific topics regarding SOTA, safety and performance outcome development, clinical strategy and more.
Process and Templates Development
Proven procedure and document templates are available for all aspects of clinical evaluation including:
- Clinical evaluation procedure
- Clinical Evaluation Plan
- Clinical Evaluation Report
- Literature Search Protocol/Report
- State of the art (SOTA) structure
Gap Assessment
AKRA TEAM offers comprehensive gap assessments of clinical documentation to highlight areas of risk or improvement prior to submission. AKRA TEAM will rank risks from high to low while offering mitigations to close any identified gaps.
Implementation
AKRA TEAM has an experienced team of medical writers, clinicians and regulatory experts available to write clinical evaluations and related documentation for all device categories and risk-classes. We offer custom tailored solutions or comprehensive solutions to addresses the needs of any manufacturer. Additionally, if the clinical evaluation is currently under review we can provide targeted updates based on any open deficiencies.
Continuous update of documentation
AKRA TEAM additionally offers solutions for continuous documentation updates. AKRA TEAM will schedule and revise documentation at defined intervals to ensure regulatory compliance is maintained according to the expectations of EU MDR.
Interested in our services?
Lorem ipsum dolor sit amet, consec tetur adipis cing elit. Ut elit tellus, luctus nec ullam corper mattis, pulvinar dapibus leo.