一般背景
欧盟医疗器械法规(EU MDR)规定,临床研究是指在医疗器械投放市场前,对其安全性和性能开展临床研究以收集相关数据的过程。其中还包括根据欧盟MDR法规进行的重大变更和CE初始认证,其中临床研究数据被认为是解决临床证据差距的必要因素。此类研究是欧盟监管流程的重要组成部分,对于某些等级的医疗器械必须予以实施,确保其满足安全性和性能相关标准。
欧盟MDR法规临床研究的重要性更胜以往。特别是由于基于等效性的符合性评估往往不再可行,医疗器械制造商必须收集器械的临床数据,以成功实现符合性评估。
III类器械和可植入器械数据通常来源于发起人责任范围内的临床研究。这也适用于具有新预期用途或采用新技术的低等级器械(可能存在安全隐忧或其他问题)。欧盟MDR法规规定,无论所属分类如何,都应对存在安全隐忧或具有高度创新性的器械开展临床研究。
开展临床研究前,研究发起人必须获得所在欧盟成员国主管机关的许可。发起人还必须获得伦理委员会的伦理批准,该委员会负责审核研究方案,旨在确保研究符合伦理要求,同时保障研究参与者的权益和安全。
欧盟MDR法规规定,开展的临床研究必须符合良好临床实践(GCP)指南,这为临床试验的设计、开展和报告提供了一整套国际标准。该指南旨在确保临床试验中能够保障研究参与者的权益、安全及健康,并且生成的数据足够可靠,能够用于支持监管决策过程。
Supporting Subheading
An Attractive Call to Action Text
临床研究完成后,发起人必须准备一份临床研究报告(CIR),用于总结研究成果并提供医疗器械安全性和性能结论。随后,将CIR提交给主管机关进行审核,作为符合性评估流程的组成部分。
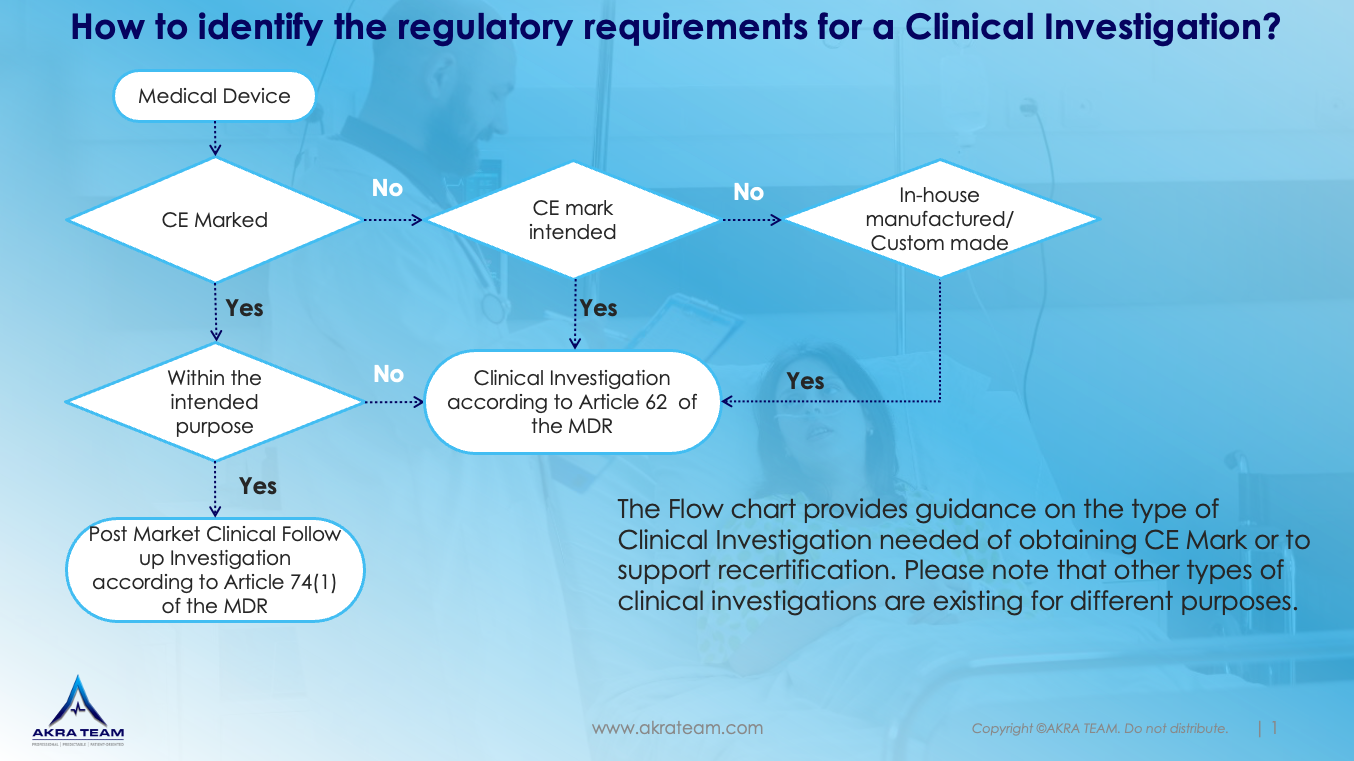
因此,规划并开展临床研究通常需要大量资源,并且取决于临床研究的适用类型,还必须满足不同的要求(参见流程图)。Akra Team可以帮助您合理设计和规划临床研究。
Key considerations
制造商需要引起注意的主要变更:
保证临床研究策略与营销策略的一致性:您计划提出的产品声明是什么?有哪些数据支持?
生成充足数据,在器械安全性和性能方面赢得利益相关者的信任。
在计划临床研究成果参数时,确保考虑风险管理数据。
考虑您所在市场的报销要求和其他潜在目标市场的监管要求。
即使部分活动采用外包模式,也应确认您的QMS系统已涵盖临床研究。
满足所在国家针对临床研究申请和文档的具体要求。
我们的服务
培训
AKRA TEAM针对欧盟MDR法规规定的临床研究一般监管要求以及医疗器械协调小组(MDCG)发布的相应指导文件提供培训。
AKRA TEAM针对ISO 14155标准“人体用医疗器械临床研究 – 良好临床实践”提供培训。
流程和模板开发
AKRA TEAM可以协助您创建临床研究程序和相关OMS程序(例如临床评估计划)以及模板。
差距评估 / 指南
AKRA TEAM可以对您的现有数据和/或临床研究计划执行差距分析,识别缺失信息并提出缩小差距的最佳方式。
AKRA TEAM可以协助您的公司确定适合当前产品的临床策略。
AKRA TEAM确保您的临床研究设计符合公认的国际指导文件,例如国际标准ISO 14155“人体用医疗器械临床研究 – 良好临床实践”以及世界医学学会发布的新版“赫尔辛基宣言”中的伦理原则(这也是MDR法规第1(64)条的要求)。
实施
AKRA TEAM提供医疗研究策略、方案开发、CRF设计以及知情同意书设计的反馈意见。在向主管机关和伦理委员会申报试验的过程中,以及在现场启动、研究启动和收尾活动中,我们也能提供支持。
文档持续更新
实施符合欧盟MDR法规要求的文档并呈递给公告机构后,为满足上市后监管等相关要求,或鉴于监管机构、市场、器械数据或制造商引起的其他变更,还需要持续更新此类文档。AKRA TEAM将开展跟进并协助制造商保持文档的合规性。