欧盟医疗器械法规(MDR)规定的临床评估
I am text block. Click edit button to change this text. Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis.
一般背景
临床评估是一个系统性且有计划的过程,用于持续生成、收集、分析并评估与器械相关的临床数据,以证明其安全性和性能,包括器械在预期用途中的临床疗效。临床评估必须符合欧盟医疗器械法规(MDR)2017/745第61条和附录XIV的要求。
这一流程包含评估可用临床证据,例如临床研究、临床文献及临床前数据,以证明器械在预期用途中安全有效,并符合当前技术水平(SOTA)。临床评估报告是总结临床评估流程结果的文档,用于支持器械实现欧盟MDR合规。报告必须在器械的整个生命周期内定期更新,以确保器械始终符合欧盟MDR法规要求,并作为器械整体技术文档的组成部分。
临床评估过程通常涵盖以下关键文档:
技术文档所含内容如下:
- 临床评估程序
- 临床评估计划(CEP)
- 针对当前技术水平(SOTA)和器械的文献搜索规范与报告
- 临床评估报告(CER)
临床评估程序描述了制造商为满足欧盟MDR法规和最新指南要求定义的相关流程和责任。
需要采取以下步骤开展临床评估:
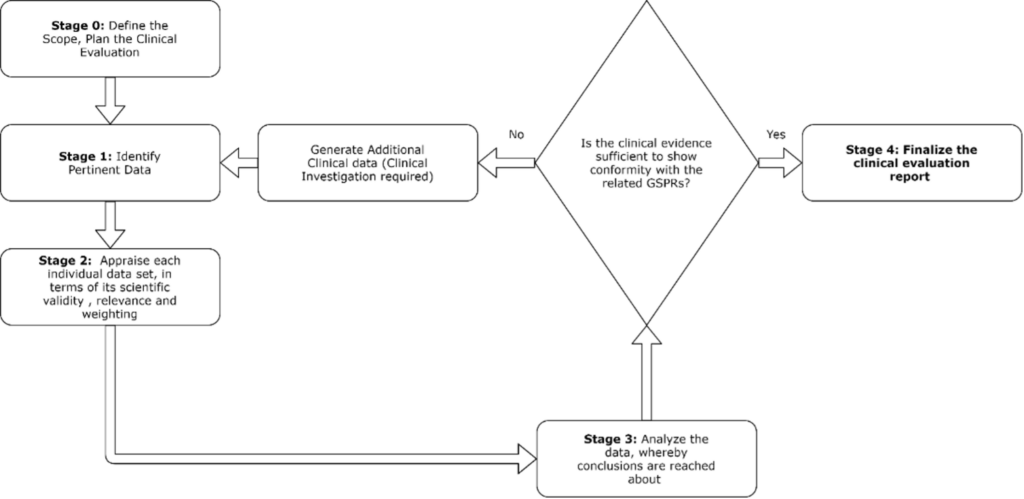
临床评估计划作为临床评估报告的基础,描述了相关数据输入和预期的文档成果。
临床评估计划包含以下内容:
- 一般安全性和性能相关要求(GSPR),需要相关临床数据的支撑
- 预期用途陈述
- 预期目标患者群,清晰注明
- 适应症和禁忌症
- 患者预期临床疗效,包含安全性和性能测量参数
- 用于评估临床安全性的定性和定量方法,明确提及测定的剩余风险和副作用
- 使用参数列表并根据药物当前技术水平确定每种适应症、预期用途或器械用途的效益-风险比接受程度
- 描述如何解决与特定成分(例如使用药物、不可存活的动物或人体组织)相关的效益-风险比问题
- 临床开发计划
临床评估报告中归纳了阶段0至3,为临床证据奠定了基础,证明器械符合一般安全性和性能相关要求。
从多个来源中识别临床证据。临床评估相关数据由制造商或第三方保存,或者也可从科学文献中查阅,用于相关器械或类似器械。必须识别与目标器械相关的临床数据。
根据临床评估计划中提出的策略识别临床数据。具体而言,按照文献搜索规范(包含相关文献数据库、搜索词、纳入和排除标准)来识别临床文献。
在评估阶段,制造商应提供数据评估方法的简短摘要,包括描述如何确定来自给定研究报告或其他来源的数据满足纳入临床评估所需的质量和相关性要求。另外还包括研究设计、偏差来源、同行评审、目标器械相关性等标准的评估。
相关数据集应在科学质量和与受试器械临床评估范围和目标的相关性基础上进行权衡。评估的接受程度应涉及以下方面:
- 妥善检索和评估文章的方法学质量和科学有效性
- 确定并记录与临床评估相关的信息
- 根据系统性标准权衡每个数据集对临床评估的作用
分析阶段旨在就临床评估报告中的临床数据充分性提供结论,以解决目标器械的一般安全性和性能要求。此外还使用证据支持临床声明,并呈现了效益-风险比结论。
根据目标器械可用评估数据集的充分性得出结论,以共同证明器械在预期用途中的安全性、临床性能和/或有效性。
效益-风险比结论应包含以下内容:
- 临床疗效摘要(涉及有意义且可衡量的患者相关临床效果)。对患者管理或公共卫生产生的积极影响。
- 临床相关性风险(例如临床数据的不确定性或限制条件、非预期副作用、滥用可能性等),并提供简要描述(例如事故、严重性、持续时间、易感患者亚组、剂量-反应关系等)。
- 风险对临床疗效的影响,考虑到所描述的因素,特别是与现有临床数据相关的不确定性。
根据评估的器械和临床策略,相应地建立临床评估报告结构。总体而言,临床评估报告包含以下部分:
- 执行摘要
- 范围
- 当前技术水平
- 受评估器械的支持证据。
- 临床前数据。
- 临床文献综述。
- 临床研究数据。
- 上市后监管和上市后临床跟踪数据。
- 数据评估
- 数据分析
- 结论
必须由以下领域且具备资质(具备相关大学学位以及5年至10年专业经验)的人员或团队评估临床评估报告:
- 研究方法论
- 信息管理
- 监管要求
- 医学写作
- 器械技术及其应用
- 症状诊断和管理,通过以下方式实现:
- 器械、医疗替代方案、治疗标准和技术知识(临床背景)
必须定期检查并更新临床评估报告,确保其保持最新状态并能够反映当前可用新信息或数据。审核频率与以下因素相关:
- 器械是否会导致显著风险
- 器械是否成熟,考虑以下因素:
- 创新
- 与受评估器械相关的临床科学、材料科学或其他科学的相应变化
- 器械临床性能和临床安全性评估中的当前可信度
- 市场上目前使用的器械总数量,以及警戒系统下的预期报告率
- 中期或长期上是否存在风险、不确定性或未解决的问题(都可能会影响更新频率)
- 设 计变更或制造程序
要点
公告机构审核过程中会严格检查当前技术水平(SOTA)。应对临床背景、医疗条件、指导方针/标准、当前医疗实践、替代疗法、类似器械、基准测试器械、SOTA效益风险比分析以及安全性和性能基准进行全面审核。
必须在临床评估报告中注明目标器械的临床声明,并提供充足的临床数据支持。
应定义所有器械的临床疗效,并与合适的可衡量性能结果相关联。临床疗效必须反映器械的预期用途。
安全性和性能结果必须定量化,与患者建立相关性,基于从最新文献综述获得的基准值确定。
根据欧盟MDR法规选择正确的临床策略是关键所在。器械是否可以做出等同性声明、是否采用成熟技术、临床数据是否适用(第61(10)条)或是否有充足的目标器械,这些都会影响临床证据的质量和数量要求。
我们的服务
培训
AKRA TEAM提供培训服务,涵盖一般临床评估和临床评估报告撰写,以及SOTA、安全性和性能结果开发、临床策略等特定主题。
流程和模板开发
我们提供经过验证的流程和文档模板,涵盖临床评估的各个方面:
- 临床评估程序
- 临床评估计划
- 临床评估报告
- 文献检索协议/报告
- 当前技术水平(SOTA)结构
差距评估
AKRA TEAM提供全面的临床文档差距评估,力求在申报前重点指出风险或改进领域。AKRA TEAM将风险从高到低排序,同时提供缓解措施来弥合已确定的差距。
实施
AKRA TEAM坐拥一支资深的医学编辑、临床医生和监管专家团队,负责撰写临床评估和相关文档,涵盖所有器械类别和风险等级。我们提供定制化或综合性解决方案,以满足所有制造商的需求。 此外,如果临床评估报告当前正在接受审核,我们还能根据已知缺陷有针对性地进行更新。
文档持续更新
AKRA TEAM额外提供文档持续更新解决方案。AKRA TEAM将在规定的时间间隔内安排工作及修订文档,确保满足欧盟MDR法规预期要求,以保持合规性。